Building Blocks File¶
By default, ChemLG looks for the building blocks in a file named: building_blocks.dat
The file should contain the molecules in SMILES or InChi format. Every molecule starts on a new line with no special characters at the beginning of the line.
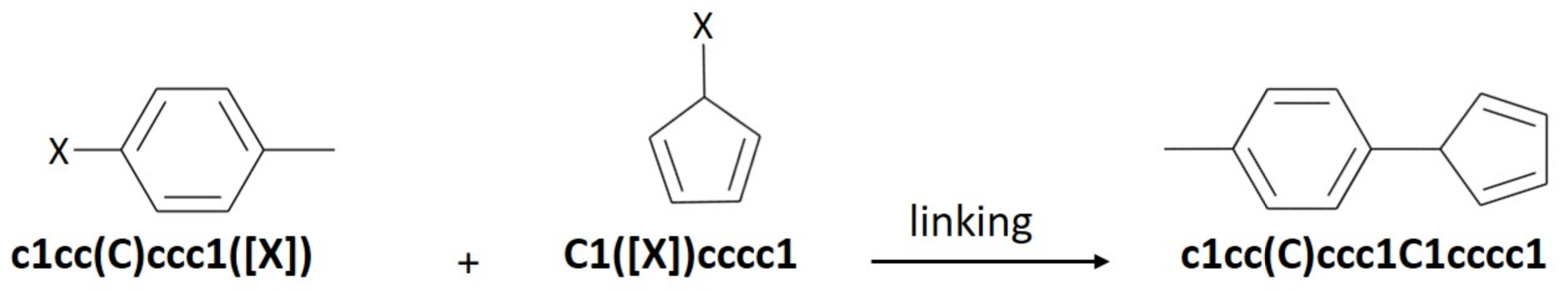
If connection is required at specific points only, add [X] to those points. If [X] is not provided, all [H] atoms will be considered for linking.
Config File¶
The config file should closely follow the template provided in the library. The rules for library generation are to be specified as:
Building Blocks: Specify the building blocks which must be present in all the molecules in the final library.
Expected type: tuple of smiles of corresponding building blocks
Default value: None
Number of Bonds: Enter the minimum and maximum values of the total number of bonds for all the molecules in the final library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Atoms: Specify the minimum and maximum number of atoms that must be present in each molecule in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Molecular Weight Range: Specify the range of the molecular weight of the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Rings: Specify the range of the number of rings present in the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Aromatic Rings: Specify the range of the number of aromatic rings present in the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Non-Aromatic Rings: Specify the range of the number of non aromatic rings present in the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Single Bonds: Specify the range of the number of single bonds present in the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Double Bonds: Specify the range of the number of double bonds present in the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Number of Triple Bonds: Specify the range of the number of triple bonds present in the molecules in the generation library (integers).
Expected type: tuple (min, max)
Default value: None
Heteroatoms: Specify the maximum number of (C, S, O, N) atoms that must be present in the molecules in the final library.
Expected type: tuple of tuple(s) ((‘Cl’, 10), )
Default value: None
Lipinski Rule: Lipinski’s rule of five is a rule of thumb to evaluate druglikeness or determine if a chemical compound with a certain pharmacological or biological activity has chemical properties and physical properties that would make it a likely orally active drug in humans. Select the choice to incorporate Lipinski rule for generating molecules in the final library.
Expected type: string (True or False)
Default value: False
Fingerprint Matching: Molecular fingerprints encode molecular structure in a series of binary digits that represent the presence or absence of particular substructures in the molecule. Comparing fingerprints will allow you to determine the similarity between two molecules. Type the target molecule and the Tanimoto index.
Expected type: comma-separated target molecules c1ccccc1-0.1, C1CCCC1-0.1
Default value: None
Substructure Inclusion: Enter substructures in SMARTS format which must be included in all molecules in the final library.
Expected type: comma separated Smiles/SMARTS
Default value: None
Substructure Exclusion: Enter substructures in SMARTS format which must be excluded in all molecules in the final library.
Expected type: comma separated Smiles/SMARTS
Default value: None
Include initial Building Blocks: Should the initial building blocks be included in the final library.
Expected type: string (True or False)
Default value: None